pytximport published
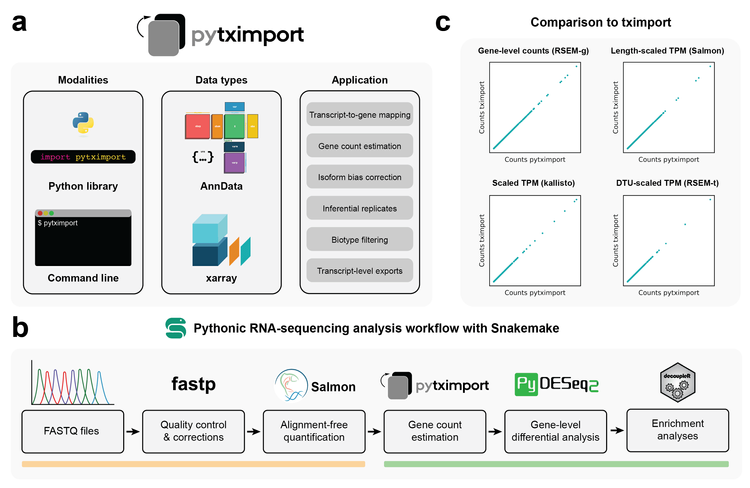
We are excited to announce the publication of our latest research paper in Bioinformatics, presenting pytximport, a Python implementation of the well-known tximport R package. This tool is designed to streamline transcript quantification in bulk RNA sequencing, addressing biases and enhancing compatibility with differential gene expression analysis tools in Python. ## What is pytximport? pytximport brings several powerful features to the table, including: - Support for a variety of input formats. - Multiple modes of bias correction. - Handling inferential replicates. - Gene-level summarization of transcript counts. - Transcript-level exports. - Generation of transcript-to-gene mappings. - Optional filtering of transcripts by biotype. ## Integration and Workflow pytximport is part of the scverse ecosystem, a collection of open-source Python packages for omics analyses. It includes both a Python and command-line interface, making it versatile for different user preferences. To demonstrate its utility, we have applied pytximport to a publicly available RNA-sequencing dataset, showcasing how it facilitates the creation of reproducible analysis workflows using Bioconda and scverse packages, all orchestrated through Snakemake rules. ## Why pytximport? By integrating pytximport into your analysis workflow, you can ensure more accurate and reliable results in RNA-seq data analysis, leveraging the robust tools provided by the scverse ecosystem. We invite you to read our paper and explore pytximport to enhance your omics data analyses. ## Links [Paper](https://academic.oup.com/bioinformatics/article/40/12/btae700/7905459) [pytximport documentation](https://pytximport.readthedocs.io/en/stable/start.html) [GitHub repository](https://github.com/complextissue/pytximport)

